install.packages(c("MatchIt","cobalt"))
library(MatchIt)
library(cobalt)0. 배경
성향점수 매칭 (Propensity Score Matching,PSM) 은 관찰 연구에서 사용되는 통계기법 중 하나로서, 무작위 대조군 연구 (Randomized control trials, RCT)가 불가능할 때 치료제나 중재(intervention)의 효과를 평가하기 위해 사용됩니다.
Note
무작위 대조군 연구(RCT)란 임상 연구분야에서 자주 활용되는 실험 방법 중 하나로서 신약이나 의료기기, 치료법과 같은 중재의 안전성과 효과성을 결정하기 위해 사용되는 방법입니다.
RCT는 관찰연구와 다르게 편향과 교란 변수들을 최소화하기 때문에, 중재와 결과의 인과관계를 평가하는 표준으로 간주됩니다.
PSM은 치료제나 중재의 효과가 결과 변수(예시. 질병의 발생, 사망 등)에 미치는 영향을 확인하고자 할 때, 그 인과관계에 영향을 미칠 수 있는 변수들의 편향(bias)를 줄이는 것을 목표로 합니다. 이런 변수들을 교란 변수라고 합니다.
PSM의 핵심은 성별, 나이 등과 같이 관측된 특성을 바탕으로 치료 또는 중재를 받을 확률인 <성향점수(Propensity score)를 계산>하는 것입니다.
이 성향점수를 계산하기 위해서는 주로 로지스틱 회귀분석이 사용됩니다. 성향점수가 계산되면, 성향점수를 바탕으로 치료를 받은 환자들과 받지 않은 환자들이 매칭되게 됩니다.
이렇게 PSM을 통해 치료를 받은 집단과 받지 않은 집단의 차이를 최대한 줄인 뒤, 치료나 중재의 인과성(causal effect)을 확인할 수 있습니다.
이번 포스트에서는 R을 활용해 PSM을 수행하는 방법에 대해 살펴보도록 하겠습니다.
1. 패키지 설치
PSM을 수행하기 위해 필요한 패키지는 MatchIt과 cobalt입니다. MatchIt은 PSM을 수행하기 위한 패키지, cobalt는 PSM 결과를 시각화하기 위한 패키지입니다.
PSM 설명을 위해 MatchIt 패키지에 있는 lalonde 데이터를 사용하겠습니다.
lalondelalonde 데이터의 변수는 다음과 같이 구성되어있습니다.
treat: 치료 집단 구분 (0: 통제 집단, 1: 치료 집단)age: 나이educ: 교육받은 연수race: 인종 (Black, Hispanic, White)married: 혼인상태 (0: 미혼, 1: 기혼)nodegree: 고등학교 미졸업 여부 (0:졸업, 1: 미졸업)re74,re75,re78: 1974, 1975, 1978년 연간소득($)
2. PSM 예시
PSM은 크게 계획 매칭 평가 세 단계로 구분됩니다. 지금부터 이들 각각에 대해 살펴보도록 하겠습니다.
1) 계획
계획 단계는 treatment에 대해 아래의 내용을 선택하는 단계입니다.
측정될 효과(effect) 어떤 변수의 효과를 평가할 것인지 결정해야 합니다. 예를 들어서 특정 약물의 사용 여부에 따른 환자들의 질병 발생이나 사망에 미치는 영향을 분석하고자 할 때, 특정 약물의 사용이라는 변수를 기준으로 층화하여 매칭을 하는 것이 필요합니다.
목표 대상 (target population) 목표 대상이란 연구 결과를 적용하고 싶은 대상을 의미합니다. 일반적으로 랜덤 샘플링한 대상에게 연구 결과를 적용하는데, 만약 샘플이 랜덤하지 않다면 목표 대상을 설정하기 어렵습니다.
- Average treatment effect in the population (ATE): 목표 대상에 있는 모든 대상들에 대한 치료제의 평균 효과
- Average treatment effect in the treated (ATT): 치료를 실제로 받은 사람들과 비슷한 사람들에 대한 치료제의 평균 효과 대부분의 매칭 방법은 ATT를 평가하는 것이 더 낫지만, 일부 매칭은 ATE도 사용 가능합니다.
균형을 맞출 공변량 (covariates)
- 공변량을 선택하는 것은 치료제의 인과관계를 타당하게 해석하기 위해 교란효과를 없애기 위한 확실한 방법이기 때문에 중요합니다. 총 인과를 평가하기 위해서 모든 공변량들은 치료제 사용 이전에 측정이 되어야 합니다. 이상적으로 공변량은 결측값(missingness)이 없어야 합니다.
우선 매칭 이전에 기존 데이터의 불균형을 확인해보겠습니다.
initial <- matchit(treat ~ age + educ + race + married + nodegree + re74 + re75, data = lalonde, method=NULL, distance = "glm")
summary(initial)
Call:
matchit(formula = treat ~ age + educ + race + married + nodegree +
re74 + re75, data = lalonde, method = NULL, distance = "glm")
Summary of Balance for All Data:
Means Treated Means Control Std. Mean Diff. Var. Ratio eCDF Mean
distance 0.5774 0.1822 1.7941 0.9211 0.3774
age 25.8162 28.0303 -0.3094 0.4400 0.0813
educ 10.3459 10.2354 0.0550 0.4959 0.0347
raceblack 0.8432 0.2028 1.7615 . 0.6404
racehispan 0.0595 0.1422 -0.3498 . 0.0827
racewhite 0.0973 0.6550 -1.8819 . 0.5577
married 0.1892 0.5128 -0.8263 . 0.3236
nodegree 0.7081 0.5967 0.2450 . 0.1114
re74 2095.5737 5619.2365 -0.7211 0.5181 0.2248
re75 1532.0553 2466.4844 -0.2903 0.9563 0.1342
eCDF Max
distance 0.6444
age 0.1577
educ 0.1114
raceblack 0.6404
racehispan 0.0827
racewhite 0.5577
married 0.3236
nodegree 0.1114
re74 0.4470
re75 0.2876
Sample Sizes:
Control Treated
All 429 185
Matched 429 185
Unmatched 0 0
Discarded 0 0PSM 이전의 treat과 control 간의 불균형을 확인할 수 있는 지표는 Standardized mean difference (Std. Mean Diff, SMD)입니다. SMD는 절댓값이 0에 가까우면 가까울수록 treat과 control 간의 균형이 맞는다고 이야기합니다. 통상적으로 연구에서 매칭이 잘 되었다(균형이 잡혔다)고 말하는 SMD의 수준은 절댓값 기준 0.1 이하입니다.
2) 매칭
이제 매칭을 진행해보겠습니다. 매칭에는 다양한 클래스들과 방법들이 존재합니다. 여기서는 1:1 nearest neighbor (NN)매칭을 통해 성향점수를 계산해보도록 하겠습니다.
NN 방법을 통해 각 treated 데이터는 가장 가까운 성향점수를 갖는 control 데이터와 짝지어게 됩니다. 짝지어지지 못한 데이터들은 매칭되지 않은 것으로 간주되어 추후 분석에서 제외됩니다.
m1 <- matchit(treat ~ age + educ + race + married + nodegree + re74 + re75,
data = lalonde,
distance = "glm",
method="nearest")
m1A matchit object
- method: 1:1 nearest neighbor matching without replacement
- distance: Propensity score
- estimated with logistic regression
- number of obs.: 614 (original), 370 (matched)
- target estimand: ATT
- covariates: age, educ, race, married, nodegree, re74, re75matchit() 에 사용되는 인자들은 크게 다음과 같습니다.
formula: 치료군과 대조군 간에 어떤 변수를 활용해 매칭을 할 것인지 작성합니다.위의 코드를 예시로 들면
treat ~ age + educ + race + married + nodegree + re74 + re75는treat를 기준으로age~re75간 변수가 비슷한 항목들을 매칭한다는 의미입니다.
distance: 치료군과 대조군 간의 거리를 측정하기 위한 방법으로, 기본값은glm입니다. 그 외에 마할라노비스 거리(mahalanobis), 유클리드 거리 등이 올 수 있습니다.methods: 매칭 방법입니다. 대표적으로nearest,optimal등이 있습니다.replace: 매칭 시 한 번 매칭된 대조군을 다른 치료군과의 매칭에도 사용할 지 여부를 결정합니다(TRUE/FALSE).caliper: 치료군과 대조군 간 매칭을 할 때 허용되는 차이를 입력합니다. 통상적으로0.2가 많이 사용됩니다.ratio: 치료군과 대조군 간의 매칭 비율입니다. 2이면 1:2 매칭, 3이면 1:3 매칭이 진행됩니다.
matching 방법 (method)
nearest: 최근접 이웃 매칭은 매칭 시 가장 많이 활용되는 방법으로, greedy 매칭이라고도 불립니다. 이는 치료군과 대조군의 쌍이 어떻게 매칭됐는지 또는 어떻게 매칭 될지 참고하지 않은 채로 매칭을 진행하기 때문입니다.따라서 최근접 이웃 매칭은 최적화와 거리가 먼 방법입니다. 이름에서도 알 수 있듯, 각 치료군과 가장 가까운 거리에 있는 대조군의 데이터를 짝짓게 됩니다. 이 때 거리가 가까운지는 성향점수의 차이를 바탕으로 판단합니다.
즉 치료군과 통제군 간의 성향점수의 차이가 가장 적은 것들을 매칭하는 것입니다.
optimal: optimal pair 매칭은 optimal 매칭이라고 줄여서 부르는데, 각 치료군과 하나 이상의 대조군을 짝지으려 한다는 점에서 nearest neighbor 매칭과 굉장히 유사합니다.그러나 최근접 이웃 매칭과 달리, greedy 하지 않고 optimal 하다는 점에서 차이가 있습니다. 이 때 optimal 하다는 것은 각 쌍의 간격의 절댓값의 합을 최적화하는 것입니다.
Note
이 외에도 full, genetic, exact 등의 매칭방법이 있습니다. 해당 매칭 방법들은 추후 작성할 예정입니다.
매칭 이후에는 matchit을 통해 만들어진 m1 에는 weights(매칭 가중치), subclass (매칭 쌍), distance (성향점수 평가), 그리고 match.matrix (treat에 매칭된 control 데이터) 등 어떻게 매칭이 되었는지 확인할 수 있습니다.
names(m1) [1] "match.matrix" "subclass" "weights" "X" "call"
[6] "info" "estimand" "formula" "treat" "distance"
[11] "discarded" "model" 3) 평가
매칭 이후에는 매칭이 잘 되었는지 평가가 필요합니다. 만약 매칭을 한 이후에도 공변량의 균형이 맞지 않다면 매칭은 실패한 것으로 간주되고 다른 매칭을 시도해야 합니다. 매칭 객체 평가 방법은 크게 두 가지가 있습니다.
1. 매칭결과 요약
첫 번째는 summary()를 통해 공변량들의 SMD 절댓값이 0.1 미만인지 확인하는 것입니다.
summary(m1)
Call:
matchit(formula = treat ~ age + educ + race + married + nodegree +
re74 + re75, data = lalonde, method = "nearest", distance = "glm")
Summary of Balance for All Data:
Means Treated Means Control Std. Mean Diff. Var. Ratio eCDF Mean
distance 0.5774 0.1822 1.7941 0.9211 0.3774
age 25.8162 28.0303 -0.3094 0.4400 0.0813
educ 10.3459 10.2354 0.0550 0.4959 0.0347
raceblack 0.8432 0.2028 1.7615 . 0.6404
racehispan 0.0595 0.1422 -0.3498 . 0.0827
racewhite 0.0973 0.6550 -1.8819 . 0.5577
married 0.1892 0.5128 -0.8263 . 0.3236
nodegree 0.7081 0.5967 0.2450 . 0.1114
re74 2095.5737 5619.2365 -0.7211 0.5181 0.2248
re75 1532.0553 2466.4844 -0.2903 0.9563 0.1342
eCDF Max
distance 0.6444
age 0.1577
educ 0.1114
raceblack 0.6404
racehispan 0.0827
racewhite 0.5577
married 0.3236
nodegree 0.1114
re74 0.4470
re75 0.2876
Summary of Balance for Matched Data:
Means Treated Means Control Std. Mean Diff. Var. Ratio eCDF Mean
distance 0.5774 0.3629 0.9739 0.7566 0.1321
age 25.8162 25.3027 0.0718 0.4568 0.0847
educ 10.3459 10.6054 -0.1290 0.5721 0.0239
raceblack 0.8432 0.4703 1.0259 . 0.3730
racehispan 0.0595 0.2162 -0.6629 . 0.1568
racewhite 0.0973 0.3135 -0.7296 . 0.2162
married 0.1892 0.2108 -0.0552 . 0.0216
nodegree 0.7081 0.6378 0.1546 . 0.0703
re74 2095.5737 2342.1076 -0.0505 1.3289 0.0469
re75 1532.0553 1614.7451 -0.0257 1.4956 0.0452
eCDF Max Std. Pair Dist.
distance 0.4216 0.9740
age 0.2541 1.3938
educ 0.0757 1.2474
raceblack 0.3730 1.0259
racehispan 0.1568 1.0743
racewhite 0.2162 0.8390
married 0.0216 0.8281
nodegree 0.0703 1.0106
re74 0.2757 0.7965
re75 0.2054 0.7381
Sample Sizes:
Control Treated
All 429 185
Matched 185 185
Unmatched 244 0
Discarded 0 0Summary of Balance for Matched Data 부분의 Std.Mean Diff 을 통해 각 변수별로 매칭이 잘 이루어졌는지 확인할 수 있습니다. 결과를 보니 일부 변수들에서 SMD가 0.1이 넘어가는 것이 보입니다. 매칭이 제대로 이루어지지 않은 것을 알 수 있습니다.
2. 매칭결과 시각화
다음으로는 그래프를 이용해 매칭 전 SMD와 매칭 후의 SMD를 비교하는 것입니다. 여기서 cobalt 패키지를 이용하게 됩니다.
love.plot(m1,
abs=T,
thresholds = 0.1,
drop.distance = T,
binary='std',
position='bottom',
var.order = 'unadjusted',
# var.names = new.name,
size = 2,
shapes=c('circle filled','circle')
)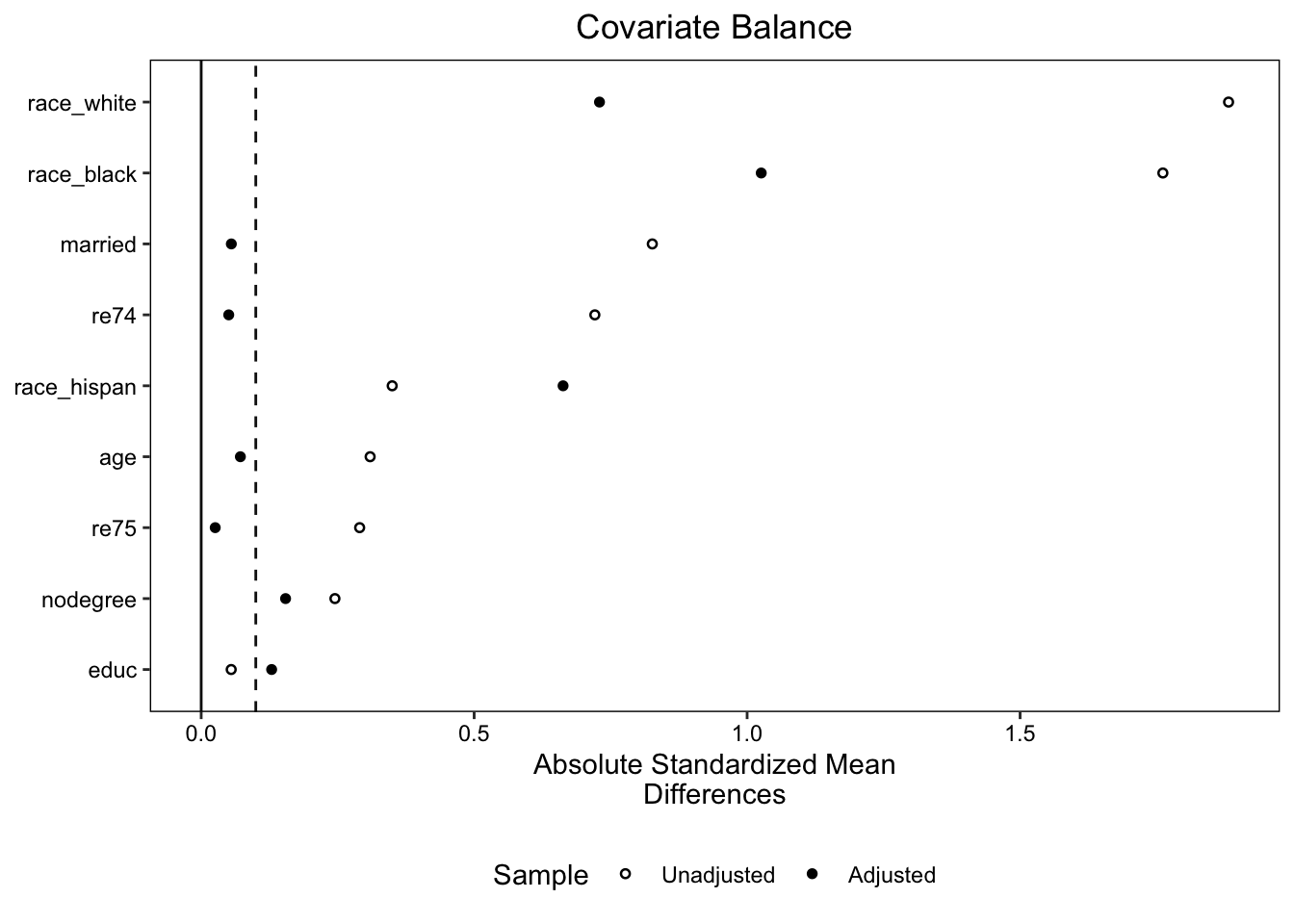
위의 그래프를 살펴보면, 매칭을 통해 adjusted된 공변량들의 SMD(검은 점)가 점선으로 나타난 0.1 이상인 경우가 꽤 많은 것을 알 수 있습니다. 매칭이 제대로 이루어지지 않은 것을 알 수 있습니다. 이럴 경우, 다른 방법으로 매칭을 시도하여 치료군과 대조군을 올바르게 매칭해주는 것이 필요합니다.
m2 <- matchit(treat ~ age + educ + race + married + nodegree + re74 + re75,
data = lalonde,
distance = "glm",
method="nearest",
caliper=.2
)
love.plot(m2,
abs=T,
thresholds = 0.1,
drop.distance = T,
binary='std',
position='bottom',
var.order = 'unadjusted',
# var.names = new.name,
size = 2,
shapes=c('circle filled','circle')
)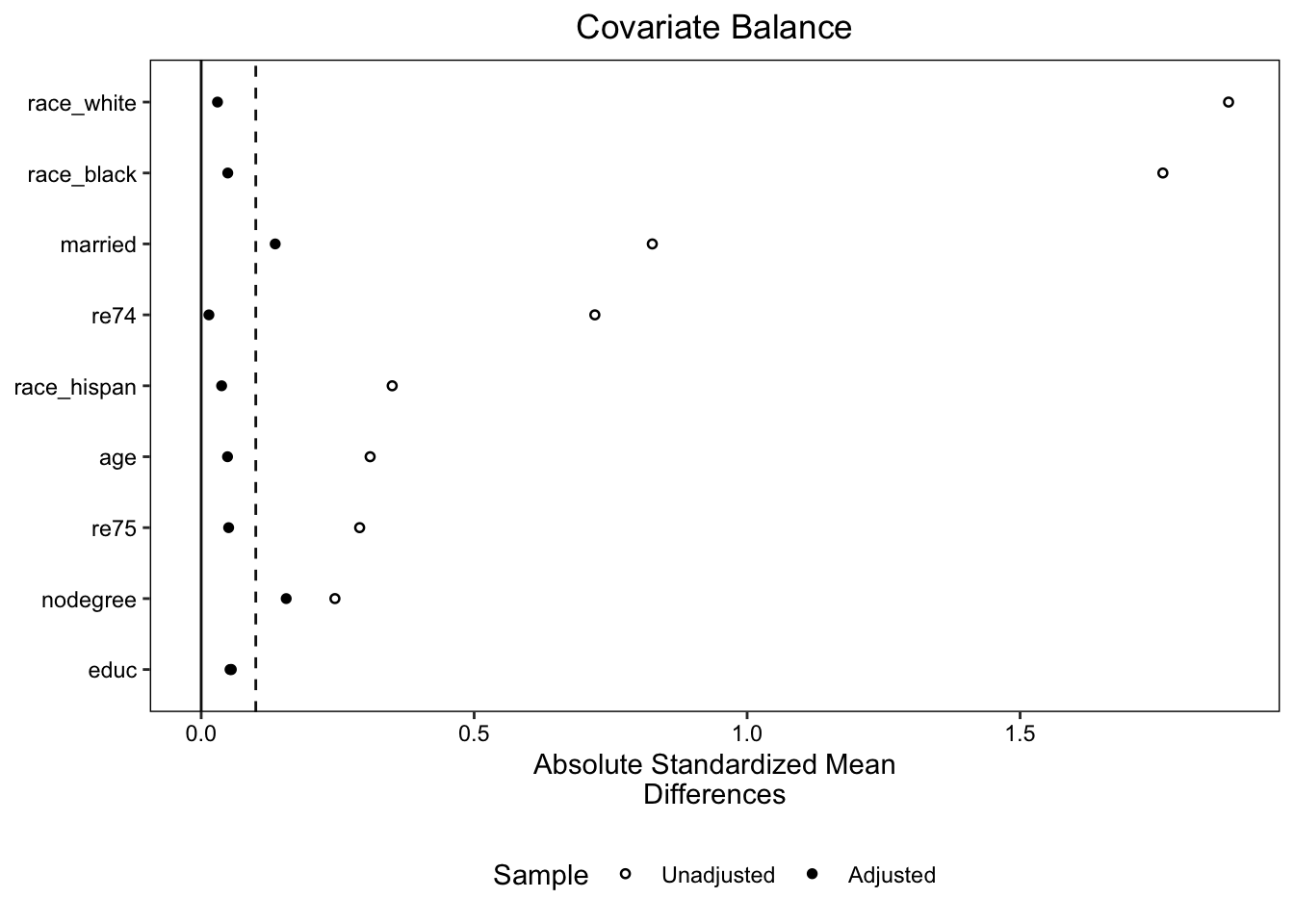
caliper를 0.2로 설정하여 매칭 범위를 넓혔습니다. 다시 말해 성향점수의 차이가 0.2보다 작은 경우는 매칭을 모두 허용했다는 뜻입니다. 그 결과, 비록 일부 변수는 여전히 SMD가 0.1 이상이지만, 첫 번째 매칭보다 SMD가 많이 줄어든 것을 알 수 있습니다.
4) 활용
MatchIt을 통해 매칭이 잘 이루어졌다면 match.data() 또는 get_matches()를 이용해 매칭된 데이터를 데이터프레임으로 만들어줄 수 있습니다. 이후 회귀분석, 생존분석 등 추가 모델링 작업을 진행해주면 됩니다.
Note
match.data()과 get_matches() 모두 매칭 이후 거리(distance), 가중치(weights), 하위그룹(subclass) 변수들이 추가된 데이터프레임을 반환합니다.
get_matches()는 매칭 시 대조군을 재사용했을 때(replace=TRUE) 활용하고, 나머지 경우에는 match.data()를 사용해 매칭된 데이터를 데이터프레임으로 변환합니다.
matched_df <- match.data(m1)
head(matched_df)참고자료
- https://cran.r-project.org/web/packages/MatchIt/vignettes/MatchIt.html
- https://ngreifer.github.io/cobalt/